理论计算材料的氧空位Oxygen Vacancies。内容涵盖的定义、形成机制、计算方法(如和)以及在催化、能源存储和光电材料中的重要性。
DOI: 10.1016/j.apcatb.2025.125413
催化活性。
22氧空位的核心作用源于缺陷工程理论,其中空位降低能带隙、产生局部电子态,从而增强光催化或电化学性能这些计算工具不仅能预测空位稳定性,还能评估其对材料热、电和光学性质的调控,推动从缺陷设计到应用优化的材料创新。
材料的氧空位的理论计算方法
。以下介绍主要计算方法及其在氧空位中的应用。
密度泛函理论(DFT)
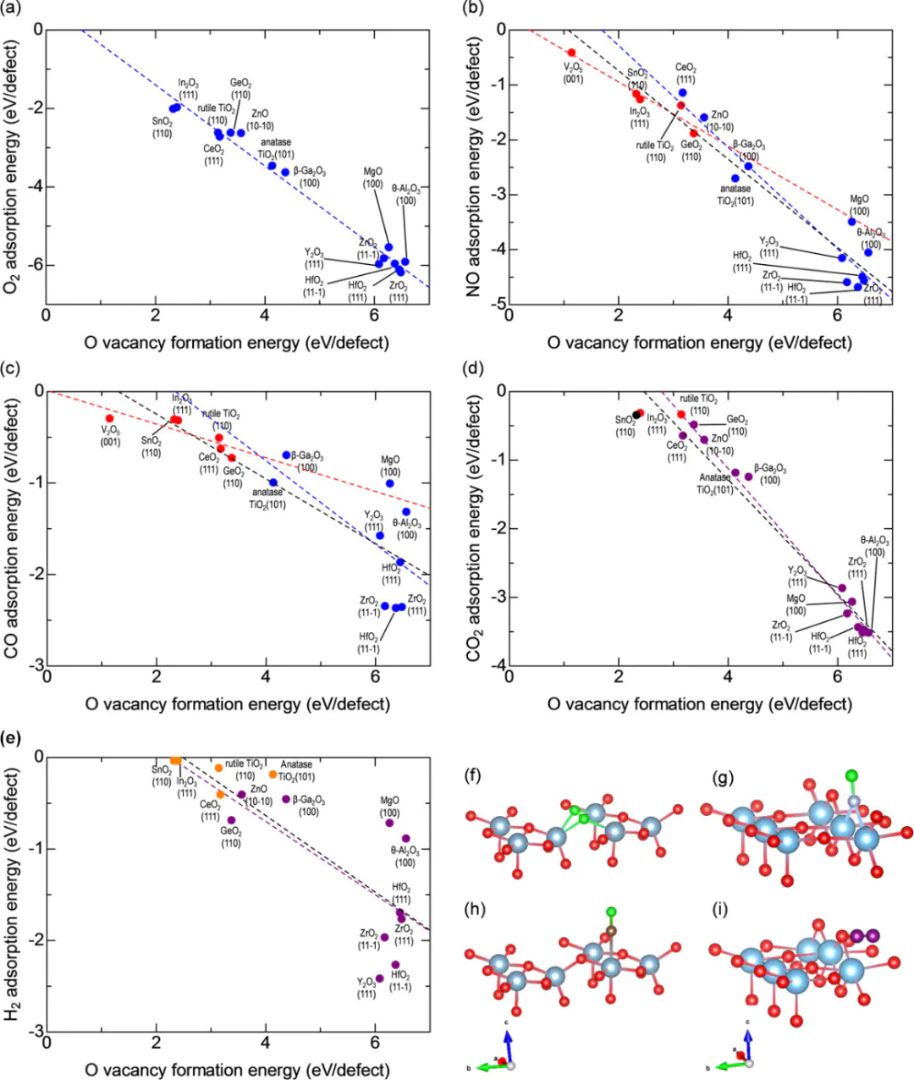
密度泛函理论基于量子力学,计算氧空位的电子结构、形成能和反应势垒,是研究氧空位最常用的方法其核心优势是无需经验参数,直接从电子密度层面预测缺陷诱导的局部态和能带变化例如,DFT用于模拟金属氧化物中的氧空位形成能(E),揭示空位如何通过电荷补偿降低体系能量(如在TiO中,E约2-4 eV),从而增强催化活性。该方法特别适用于氧化物材料,在评估空位对的影响时表现出色。
DFT在氧空位工程中应用广泛,如预测CeO催化剂中空位对CO还原的促进作用,或优化锂离子电池阴极中的离子迁移路径。例如,DFT计算显示氧空位可将能带隙从3.2 eV降至2.5 eV,提升光吸收效率。
。
分子动力学(MD)
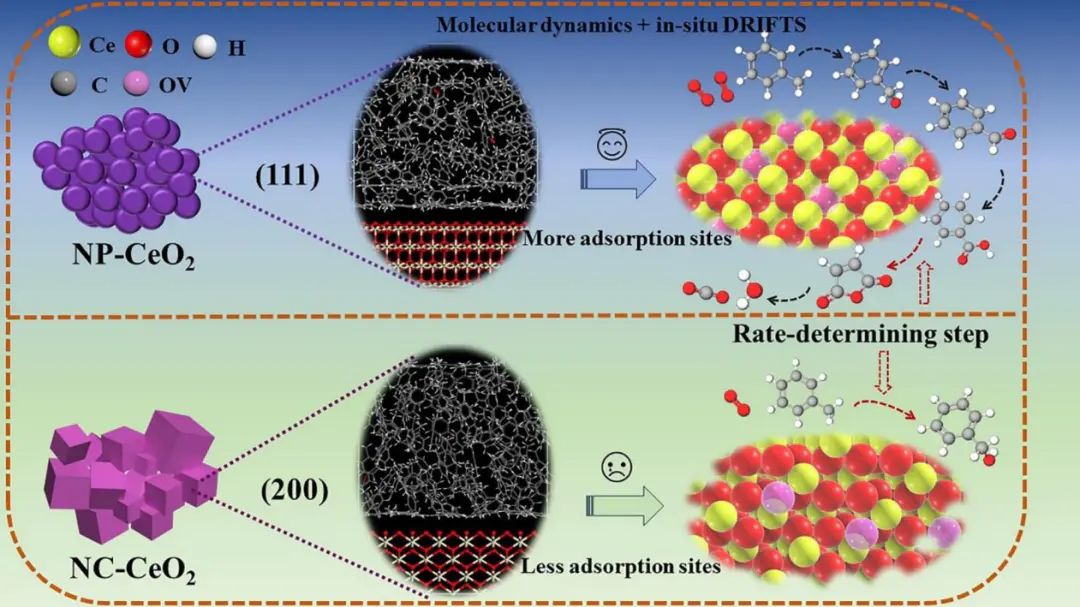
分子动力学通过经典力学模拟原子运动,研究氧空位的动态行为和有限温度效应,适用于大尺度体系的动态分析例如,分子动力学模拟直接表明,在实际反应温度下,甲苯分子更容易吸附在CeO的(111)晶面上。
3+4+应用:例如,在电池材料中,MD模拟揭示氧空位如何加速Li扩散,改善循环性能。。
机器学习
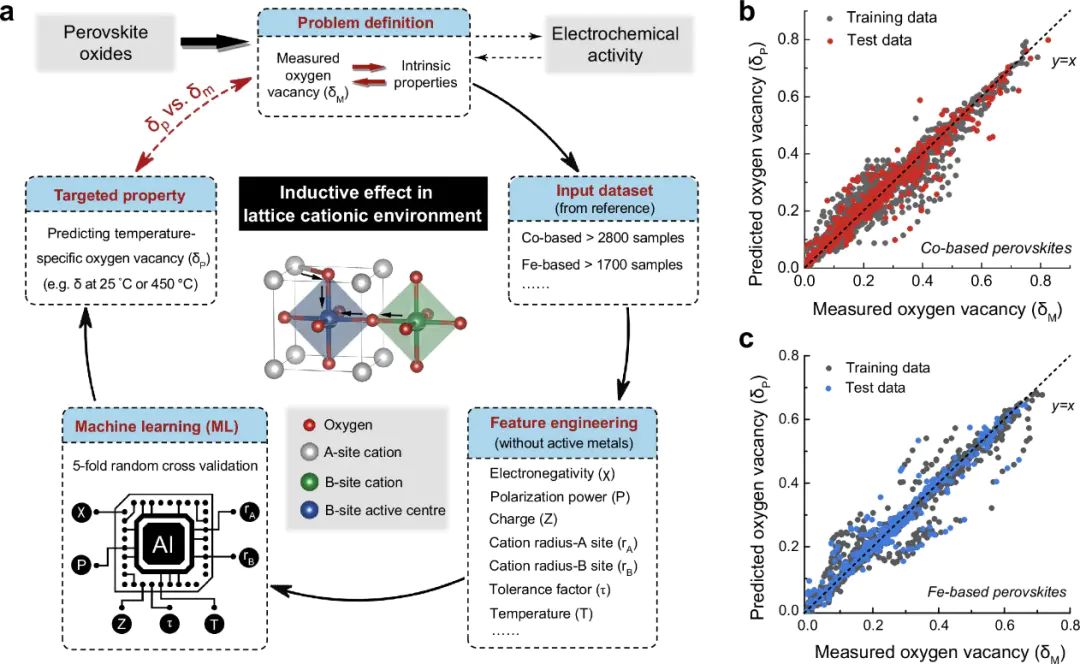
机器学习通过数据驱动方法,优化氧空位的计算效率和预测精度,特别适合高通量筛选和复杂体系分析例如,揭示了阳离子诱导相互作用在不同温度下预先确定235种钴基和200种铁基钙钛矿催化剂的氧空位浓度中的作用,这种趋势可以从基于阳离子晶格环境的机器学习技术中很好地预测,不需要大量的计算和实验输入。
机器学习指导应用:挑战在于训练数据的质量,需结合高精度DFT数据和实验验证以确保模型可靠性材料的氧空位作为缺陷工程的核心,通过调节电子结构和动态行为实现性能提升,成为材料科学和能源领域的焦点。。