常以晶相、价态、粒径、和负载方式来描述XRDXPSTEM表面原子催化剂,它可能和合成后表征得到的初始结构相差很远。
结构重构对于热催化、电催化和光催化,活性位点的几何结构、电子态和局部反应环境都会随反应条件改变,催化速率和选择性也随之改变。
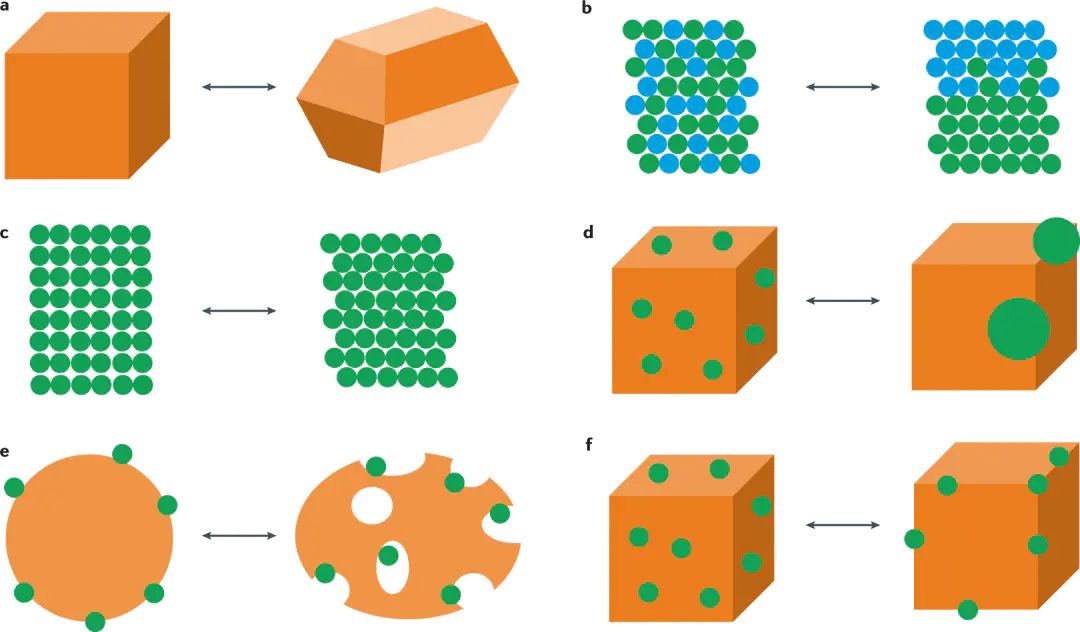
反应速率测试中的催化剂结构不能只等同于反应前样品结构温度升高会增强表面扩散,金属颗粒可能发生团聚、分散或形貌调整。电催化中,电位改变电子化学势,表面金属价态、吸附氧物种和水层取向随电位移动。
图|反应条件会驱动催化剂从初始结构进入工作态结构,结构变化影响活性和选择性。
工作态结构由材料本身和外部反应条件共同确定催化剂表面存在台阶、棱边、角位、缺陷和晶界,这些位置的原子配位数低于体相。低配位原子拥有较高表面能,也更容易与吸附物发生强相互作用。反应开始后,低配位位点常成为结构变化的起点,原子迁移会降低局部表面能或形成更适合吸附中间体的构型。
若活性评价前后只比较平均粒径,表面晶面与缺陷密度的变化容易遗漏不同晶面的原子排列、电子态密度和吸附位点分布不同,反应物会选择性稳定某些表面构型。台阶位、边缘位和金属氧化物界面往往具有更高反应活性,也更容易在反应中迁移。活性位点和结构不稳定性经常出现在同一类高能表面区域,这使催化剂性能具有动态特征。
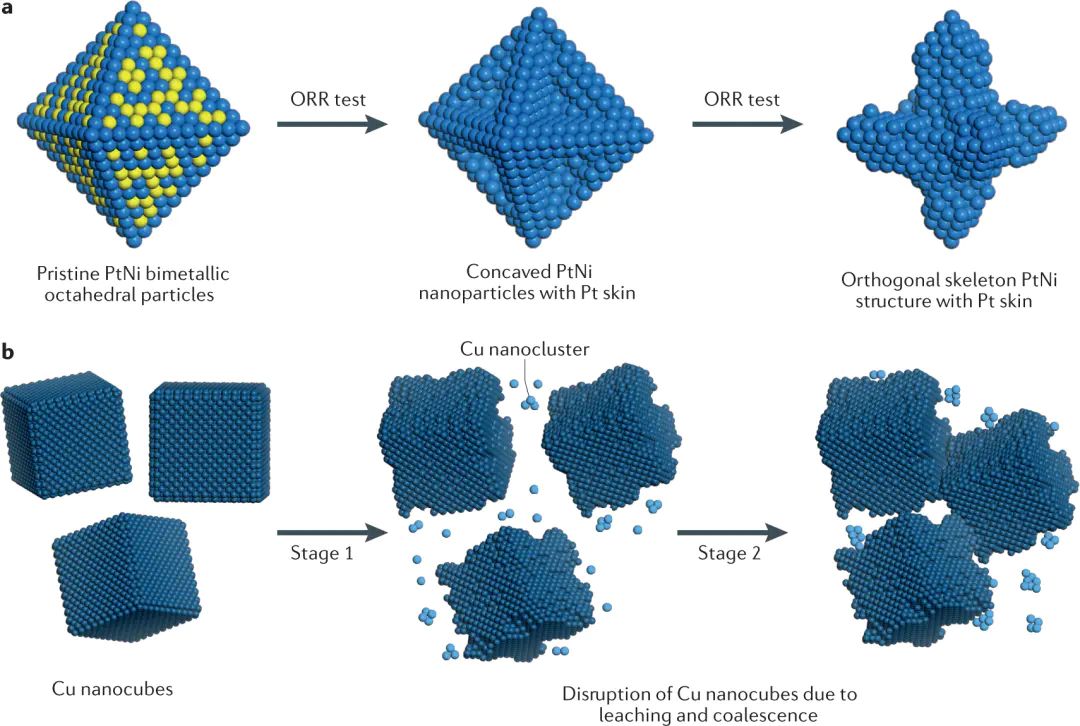
价态为什么会随反应气氛变化
氧空位和可逆氧化还原
O2H2O氧空位把表面结构变化和电子补偿过程连接在一起CeO2这类可还原氧化物具有明显的氧储放能力。转换、氧空位迁移和金属颗粒形貌变化可以同时发生。反应速率提高时,表面并非只多了几个吸附物,金属载体界面、氧迁移路径和局部价态都会参与反应循环。
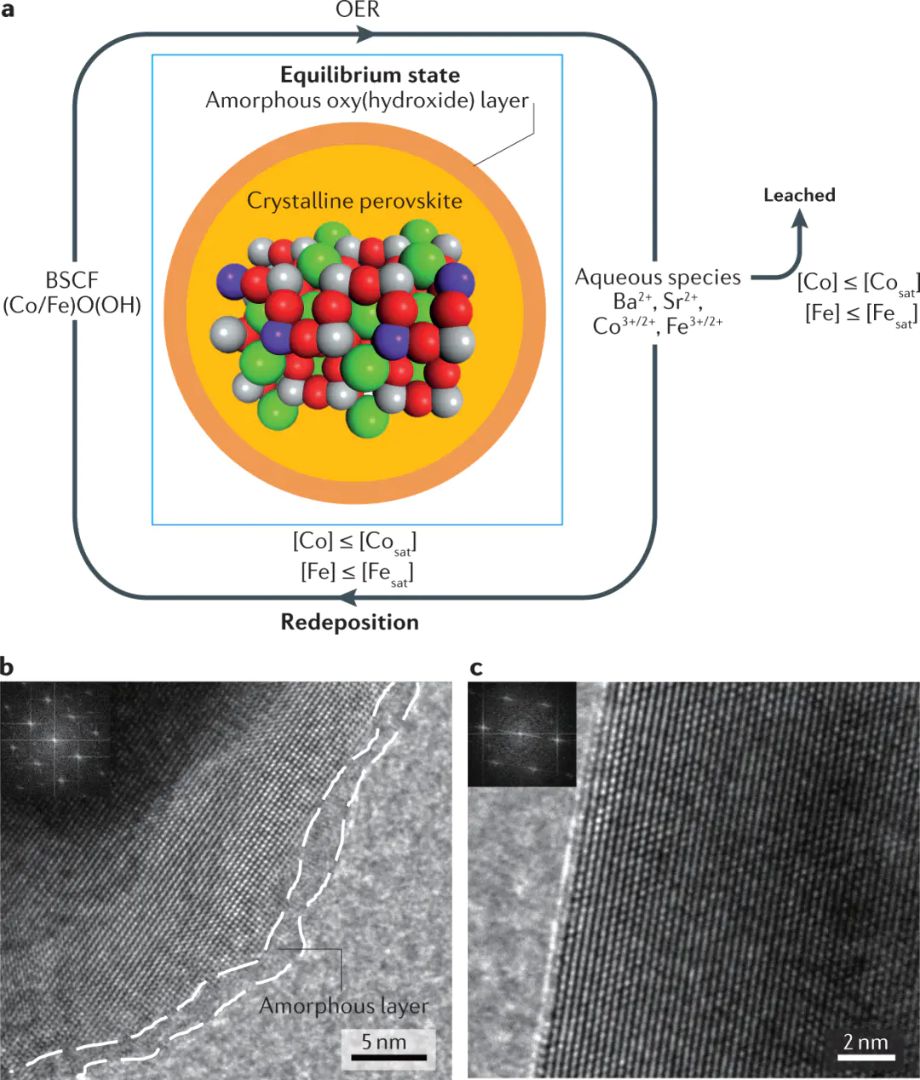
XPSXASRamanTEM只用反应后离线表征很难完整捕捉这些可逆过程析氧反应需要水或在表面吸附、脱质子、形成、、等中间体。强氧化电位会推动金属位点升高价态,表面逐步羟基化、氧化或非晶化。。
OER图|钙钛矿氧化物在中常形成表面羟基化、金属价态变化和氧化层重构。
预催化剂与工作态催化剂
CoNiFeM—O图|在条件下可转化为参与反应的钴氧羟化物类表面结构。
若把活化后的电流全部归因于初始晶相,结构活性关系会出现偏差光催化材料在暗态下具有一个平衡结构,光照后电子和空穴分离并迁移到不同区域。电子富集区更容易发生还原,空穴富集区更容易发生氧化;反应物吸附和光生电荷转移叠加后,局部化学势发生改变。光照会把半导体表面的电子状态推离暗态平衡。
—光催化的工作界面由光生载流子、吸附物和固体结构共同塑造图|光催化体系在光照和反应气氛下会形成新的界面结构和电子状态。
金属—半导体界面的重排
TiO2CuAuPt因此,光催化性能不能只用暗态结构描述。原位、原位红外、光照条件下的、瞬态吸收光谱和环境显微技术可以追踪界面价态、吸附中间体、载流子寿命和颗粒结构。,否则得到的界面状态可能偏离反应过程。
如何表征重构?
原位谱学和显微结构
XASRamanTEMXRDPDF单一表征信号通常难以完整描述重构。价态变化不一定说明晶相改变,晶相不变也可能伴随表面层重排;颗粒尺寸稳定时,晶面比例、缺陷密度和界面氧物种仍可能变化。,尤其要比较反应前、反应中和反应后之间的可逆性。
活性、选择性和结构同步变化
—催化剂重构并不必然有害。某些重构会暴露高活性位点、增加缺陷、改善界面电荷转移;某些重构会导致烧结、溶出、相分离或活性层脱落。重构的影响取决于新形成结构是否能稳定维持反应所需位点和传输通路。
“”“”参考文献:;: