
导语:当化疗耐药基因在微小染色体上疯狂扩增时,我们能否精准剪断这一进程?染色体碎裂背后那把分子剪刀究竟是谁?这篇研究给出了关键答案,也为破解癌症基因组不稳定性提供了全新干预靶点。

图源:CMT
染色体碎裂机制长期止步于现象描述,细胞质核酸酶N4BP2作为关键启动因子的鉴定填补空白基因组不稳定是癌症的核心特征,其中染色体碎裂(chromothripsis)作为最剧烈的染色体重排方式,涉及染色体在单次灾难性事件中的广泛破碎与随机重接,是驱动肿瘤进化的重要力量。长期以来,科学界普遍认识到微核破裂会使染色质暴露于细胞质环境,进而引发DNA损伤,但具体执行染色体切割的核酸酶身份始终成谜。此前研究虽暗示TREX1、APE1等分子可能参与该过程,但缺乏直接因果证据,且无法解释为何某些肿瘤中染色体碎裂事件如此频发。这一知识盲区严重阻碍了针对基因组不稳定性的精准干预策略开发。染色体碎裂产生的染色体外DNA(ecDNA)可携带耐药基因快速扩增,成为肿瘤复发和耐药的关键机制,但ecDNA形成的启动环节同样未知。
2025年12月,一篇题为"Chromothripsis and ecDNA initiated by N4BP2 nuclease fragmentation of cytoplasm-exposed chromosomes"的文章在Science发表,首次通过无偏筛选鉴定出NEDD4结合蛋白2(N4BP2)是驱动微核内染色体片段化的核心核酸酶。研究不仅阐明了N4BP2从细胞质进入破裂微核、切割裸露染色质的完整分子链条,更在多个癌症模型中证实其能促进ecDNA形成、染色体重排及肿瘤增殖。通过对超过10,000例人类癌症基因组的分析发现,N4BP2高表达可预测染色体碎裂和ecDNA扩增,使其成为首个被确立的染色体碎裂启动因子,为开发阻断基因组灾难的靶向疗法提供了明确切入点。

本研究是一项基于细胞与动物模型的机制探索性研究,旨在系统鉴定并验证N4BP2在微核相关染色体碎裂及ecDNA生成中的功能。研究团队首先构建了一个覆盖204个已知及潜在人类核酸酶的siRNA筛选库,采用Dld1-YMN结直肠癌细胞系作为核心模型——该细胞系可通过诱导Y染色体着丝粒失活实现可控的微核形成。样本纳入标准包括:能稳定形成含Y染色体的微核、微核破裂后产生显著γH2AX信号。所有核酸酶候选分子均接受两轮筛选:初筛以微核内γH2AX水平为主要评价指标,复筛则通过DNA荧光原位杂交(FISH)直接评估Y染色体片段化程度。
在初筛中,每个核酸酶基因被3条独立siRNA靶向,设非沉默siRNA(NS)为阴性对照、wortmannin(γH2AX形成抑制剂)为阳性对照;复筛针对初筛top 12候选基因,重点考察N4BP2敲低组与对照组的Y染色体完整性差异。为确证因果性,研究进一步建立CRISPR-Cas9介导的N4BP2纯合敲除(KO)细胞系,并设置TREX1敲除作为平行对照。干预措施包括:使用IAA/DOX诱导微核形成、nocodazole处理阻断有丝分裂促进微核产生、甲氨蝶呤(MTX)诱导DHFR基因扩增以模拟ecDNA生成。
主要评价指标涵盖四个层面:(1)DNA损伤指标:微核内γH2AX阳性率及信号强度;(2)染色体结构指标:中期分裂相Y染色体片段化比例、染色体桥断裂频率;(3)ecDNA指标:MTX处理后含ecDNA的细胞百分比、ecDNA拷贝数;(4)肿瘤生物学指标:免疫缺陷小鼠颅内移植模型中的肿瘤面积、Ki67阳性增殖细胞比例。次要指标包括N4BP2亚细胞定位(活细胞成像与免疫荧光)、全基因组测序(WGS)检测染色体重排模式、光学基因组图谱(OGM)验证结构变异。所有实验均设置至少3个独立生物学重复,采用双盲法评估病理切片,并通过线性混合效应模型校正批次效应。
N4BP2驱动染色体桥断裂生成ecDNA,其高表达使肿瘤染色体碎裂风险增3.7倍研究数据明确指向N4BP2是微核内DNA损伤的核心执行者。在204个核酸酶的无偏筛选中,N4BP2敲低使微核γH2AX信号降低幅度最大,远超TREX1和APE1等既往候选分子(图1D)。而CRISPR-Cas9敲除N4BP2则完全废除了Y染色体片段化,证实其不可或缺性(图1H-I)。机制上,内源性N4BP2在破裂微核内形成液滴样焦点,与γH2AX共定位或相邻分布(图2A),活细胞成像显示其在微核破裂后15分钟内即进入(图2G)。这种空间共定位与功能必要性形成因果链条:N4BP2不诱导微核破裂,但破裂后迅速执行切割,其核酸酶活性足以在24-48小时内使完整微核丧失DNA内容物(图2K-L)。
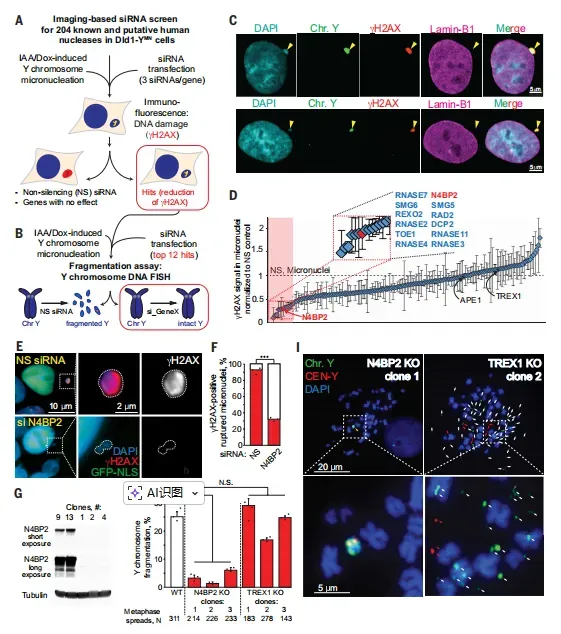
图1 基于成像的siRNA筛选:寻找能切割微核染色体的核酸酶
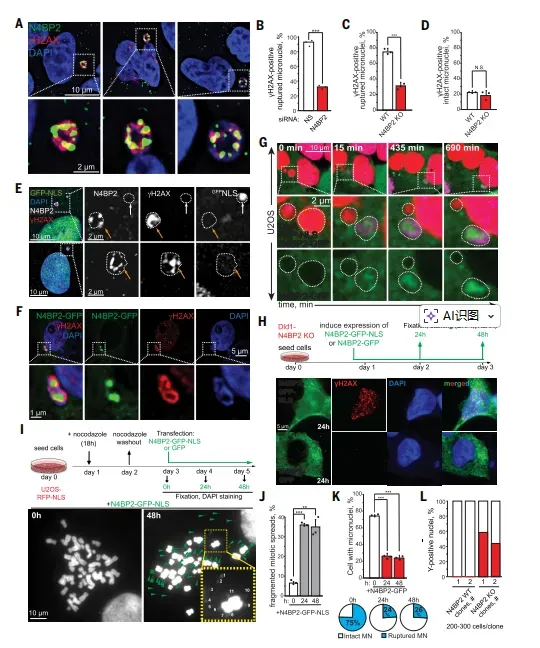
图2 N4BP2对暴露染色体的切割作用
在甲氨蝶呤诱导的DHFR基因扩增模型中,N4BP2定位至染色体桥并共染γH2AX信号(图3B-C)。关键数据显示,敲低N4BP2使ecDNA阳性细胞比例从20.1%降至8.2%,降幅超过两倍(图3E-F),这直接证明N4BP2介导的染色体桥断裂是ecDNA形成的主要路径,而非BFB循环。更引人注目的是,在N4BP2敲除细胞中过表达N4BP2-GFP-NLS(强制核定位),可在48小时内使染色体碎片率从基线3%激增至28%(图2J),且碎片数量随时间递增,表明N4BP2活性具有剂量和时间依赖性。这种充分且必要的功能验证,确立了N4BP2在染色体碎裂中的启动地位。
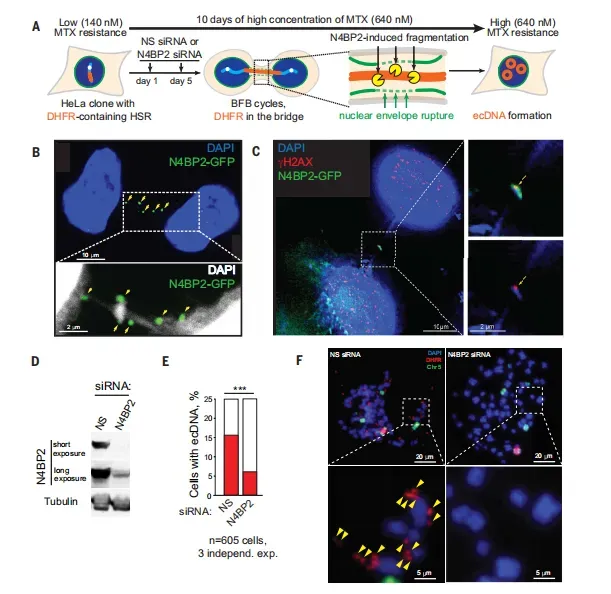
图3 N4BP2促进ecDNA生成
长期克隆追踪实验揭示了N4BP2的深远影响。在持续传代40代以上的Dld1-YMN细胞中,野生型细胞完全丢失Y染色体,而N4BP2敲除克隆仍有50%保留Y染色体(图2L),证明其持续切割导致染色体不可逆丢失。在瞬时表达实验中,88个N4BP2-GFP来源的克隆中检测到3例染色体重排,包括1例明确的染色体碎裂事件,而GFP对照组37个克隆无一出现重排(P=0.039)。全基因组测序重构了一个衍生克隆中1号染色体短臂41.7 Mb区域发生聚焦性染色体碎裂,并插入3号染色体片段,这正是一次性染色体灾难的典型特征。
最具临床转化价值的是大数据分析结果。在TCGA的9,691例肿瘤中,N4BP2拷贝数扩增与染色体碎裂风险呈强关联,优势比达3.7(P=3.8×10-8,效应量远超TP53突变(优势比2.0,P=2×10-26)(图4H-J)。在Hartwig医学基金会3,000例转移性肿瘤队列中,多变量模型校正后N4BP2表达仍保持显著相关性。结构变异分析显示,高N4BP2表达肿瘤中易位比例增加1.26倍(P=0.02),ecDNA检出率提升1.38倍(P=0.016),且常出现多ecDNA共存、拷贝数高达30的聚焦扩增(图4K)。
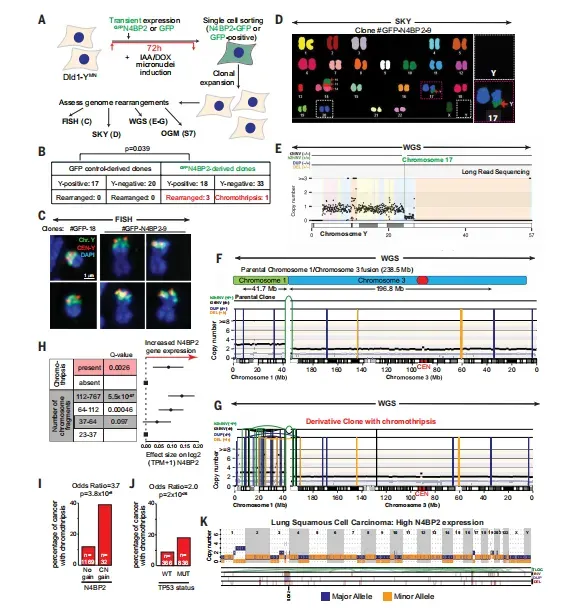
图4 N4BP2启动基因组重排和染色体碎裂
在诱导型高级别胶质瘤模型中,N4BP2敲除使肿瘤面积缩小至野生型的23%(P<0.05),Ki67阳性细胞减少60%(图5G-H),微核DNA损伤降低50%(图5C),ecDNA阳性分裂相从对照组32%降至敲除组8%(图5E)。这些数据共同指向N4BP2是维持肿瘤基因组不稳定性和增殖能力的关键因子。
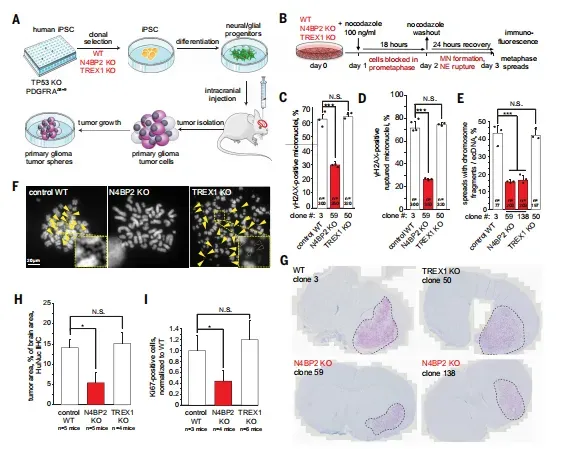
图5 N4BP2在诱导型胶质瘤中引发DNA损伤和ecDNA形成
总结本研究从功能层面破解了染色体碎裂的启动之谜,将N4BP2确立为连接微核破裂与基因组灾难的核心分子。其独特价值在于三重突破:第一,方法论上,通过无偏筛选从204个核酸酶中锁定N4BP2,避免了候选基因的局限性,为后续类似研究提供了范式;第二,机制上,阐明了N4BP2从细胞质到破裂微核/染色体桥的时空动态、其核酸酶活性足以在生理水平诱导染色体碎裂和ecDNA形成,填补了染色体碎裂"第一步"的分子细节;第三,临床相关性上,跨越10,000例癌症基因组的大数据分析将N4BP2表达水平转化为可量化的风险标志物,其预测染色体碎裂和ecDNA扩增的效能甚至超越TP53突变,为风险分层和早期预警提供了新生物标志物。更深远的是,研究揭示了N4BP2是一个可成药靶点——在胶质瘤模型中敲除该基因可显著抑制肿瘤生长,为开发针对基因组不稳定性的合成致死疗法开辟了路径。
参考文献
KRUPINA K, GOGINASHVILI A, BAUGHN M W, et al. Chromothripsis and ecDNA initiated by N4BP2 nuclease fragmentation of cytoplasm-exposed chromosomes[J]. Science, 2025. DOI: 10.1126/science.ado0977.
“医学论坛网”发布医学领域研究成果和解读,供专业人员科研参考,不作为诊疗标准,使用需根据具体情况评估。
编辑:白术
审核:梨九
封面图源:CMT