
导语:当ALK抑制剂让肺癌患者看到长期生存希望时,为何仍有超过半数患者在两年内复发?问题或许不在癌细胞本身,而在于它们周围的“邻居”——那些曾被认为“无辜”的基质细胞。最新研究揭示,这些“邻居”正通过双重信号通路暗中保护癌细胞,让靶向治疗功亏一篑。

图源:CMT
ALK抑制剂耐药机制迷雾重重,肿瘤微环境双重信号通路揭示破解之道当前,以阿来替尼(alectinib)为代表的间变性淋巴瘤激酶(ALK)酪氨酸激酶抑制剂(TKI)已将EML4-ALK融合阳性非小细胞肺癌(NSCLC)患者的五年生存率提升至60%以上,成为精准肿瘤治疗的典范。然而,临床实践中仍有30%-40%的患者在治疗初期即表现出原发性耐药,而几乎所有获得性缓解的患者最终都会面临疾病进展的困境。既往研究虽已揭示多种耐药机制——包括ALK激酶域二次突变、EGFR或KRAS等旁路信号激活、上皮-间质转化(EMT)及组织学类型转化——但仍有相当一部分耐药病例无法用上述机制解释,提示存在未被阐明的关键因素。
近年来,肿瘤微环境(TME)在耐药中的作用日益受到重视。癌症相关成纤维细胞(CAFs)作为TME中最丰富的基质成分,可通过分泌生长因子、细胞因子及重塑细胞外基质(ECM)等方式影响肿瘤生物学行为。然而,现有研究多聚焦于可溶性因子的旁分泌作用,对细胞-细胞直接接触介导的近分泌信号缺乏系统性评估。更关键的是,CAFs与肿瘤细胞之间的交互对话具有高度动态性和复杂性,传统实验方法难以同步解析两种信号类型的贡献权重。

2025年12月,Science Signaling发表了一篇题为"Cancer-associated fibroblasts confer ALK inhibitor resistance in EML4-ALK–driven lung cancer by concurrent integrin and MET signaling"的文章,首次采用创新的细胞类型特异性蛋白质组学技术,同时追踪了CAFs来源的肝细胞生长因子(HGF)-MET旁分泌轴与纤维连接蛋白(FN1)-整合素β1(ITGB1)近分泌轴在ALK-TKI耐药中的协同作用。该研究不仅填补了领域空白,更提出了双重阻断的联合治疗策略,为克服CAFs介导的耐药提供了可直接转化的临床路径,其方法论创新对解析其他肿瘤类型的基质-上皮交互机制亦具有重要借鉴意义。
创新双标记蛋白质组学技术,精准解析癌-基质细胞互作网络本研究是一项基于体外细胞模型与体内动物实验的转化医学研究,旨在系统阐明CAFs介导EML4-ALK阳性NSCLC细胞对ALK抑制剂耐药的分子机制,并评估联合阻断关键信号通路的体内疗效。研究团队首先建立了包含五种EML4-ALK融合阳性NSCLC细胞系(H3122、H2228、STE1、CUTO8、CUTO9)与四种成纤维细胞系(两种原代肺CAFs、正常肺成纤维细胞MRC5及永生化肺成纤维细胞Wi38-VA13)的多维度共培养体系,通过活细胞成像系统动态监测荧光标记肿瘤细胞的增殖活性。
实验设计包含三个递进层次:其一,比较肿瘤细胞单培养、成纤维细胞条件培养基(CM)处理及物理共培养三种模式对四种ALK抑制剂(克唑替尼、塞瑞替尼、布格替尼、阿来替尼)敏感性的影响,明确细胞接触依赖性信号的贡献;其二,采用细胞类型特异性氨基酸前体标记(CTAP)结合串联质量标签(TMT)的定量磷酸化与表达蛋白质组学技术,在单细胞分辨率下解析共培养体系中肿瘤细胞与CAFs各自的信号网络变化。该技术通过代谢酶(赖氨酸消旋酶Lyr与二氨基庚二酸脱羧酶Ddc)的工程化表达,实现d8-D-赖氨酸与二氨基庚二酸(DAP)在特定细胞类型的选择性掺入,配合TMT标记区分药物处理组与对照组,最终获得4359个磷酸化位点与4320个赖氨酸标记肽段的定量数据;其三,在体外实验明确MET与ITGB1信号轴的协同作用后,利用EML4-ALK阳性小鼠同种移植瘤模型(mEA1)评估阿来替尼单药、阿来替尼联合MET抑制剂(capmatinib)、阿来替尼联合整合素抑制剂(cilengitide)及三药联合方案的体内抗肿瘤效果。主要评价指标包括肿瘤细胞活力(基于核荧光标记计数)、关键信号蛋白磷酸化水平(通过免疫印迹与流式细胞术检测)、移植瘤体积与重量变化,次要指标涵盖小鼠体重变化以评估治疗耐受性。此外,研究还利用临床数据库(OncoSG)分析了CAFs标志物FAP-α与ITGB1表达水平与肿瘤分期及患者生存的相关性。
研究结果:双重信号协同驱动耐药,联合阻断实现肿瘤深度缓解研究首先发现,物理共培养介导的耐药强度显著高于单纯条件培养基处理。在接近临床血清峰浓度的阿来替尼(0.625μM)作用下,H3122细胞与MRC5成纤维细胞共培养后存活率提升至单培养组的3.2倍,而条件培养基处理仅提升至1.8倍,提示细胞接触依赖性信号不可或缺(图1)。通过80种细胞因子芯片筛选,研究者锁定CAFs分泌的核心因子为肝细胞生长因子(HGF),其浓度在条件培养基中达1.1-1.3 ng/mL。外源性HGF(1.3 ng/mL)可完全模拟条件培养基的耐药保护作用,而MET抑制剂capmatinib能彻底逆转该效应。然而,capmatinib对共培养介导的耐药仅部分有效(存活率从80%降至55%),证实存在MET非依赖的第二条耐药通路(图2)。
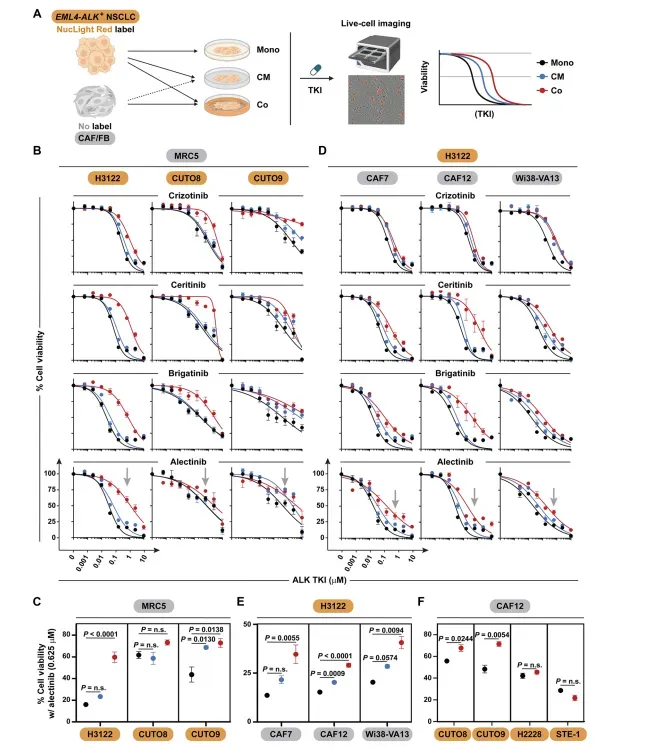
图1 成纤维细胞对EML4-ALK阳性非小细胞肺癌细胞药物敏感性的调节作用
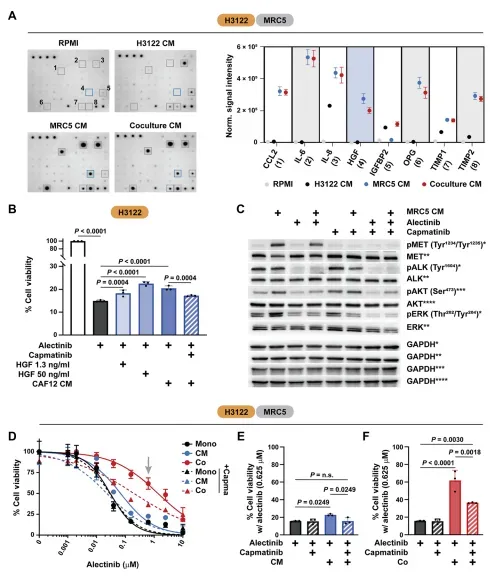
图2 成纤维细胞分泌因子对EML4-ALK阳性非小细胞肺癌细胞TKI敏感性的影响
为无偏倚地解析第二条通路,研究团队采用CTAP蛋白质组学策略。结果显示,与单培养相比,共培养诱导的磷酸化信号变化幅度远超药物本身处理效应。在阿来替尼处理的H3122细胞中,CAFs接触显著降低EGFR(Tyr1172)激活磷酸化,但增强抑制性SRC(Tyr530)磷酸化,同时特异性激活CTTN、NES、ITGA5等黏附信号蛋白。激酶底物富集分析(KSEA)预测ROCK1/2、PAK1/2、PRKCA/E等黏附相关激酶活性上调,而RSK1/2/3、AKT1等促生存激酶亦被激活。表达蛋白质组学进一步揭示,CAFs促使肿瘤细胞内FN1、多种胶原及ITGA5、CAV1表达显著上调,这些蛋白的丰度变化较磷酸化修饰对信号传导的影响更为关键。整合数据表明,CAFs通过上调ECM-整合素信号轴,特别是ITGB1异二聚体(可与ITGA2、ITGA5形成异源二聚体),代偿性激活EML4-ALK下游的ERK/RSK与AKT/mTOR通路(图3)。
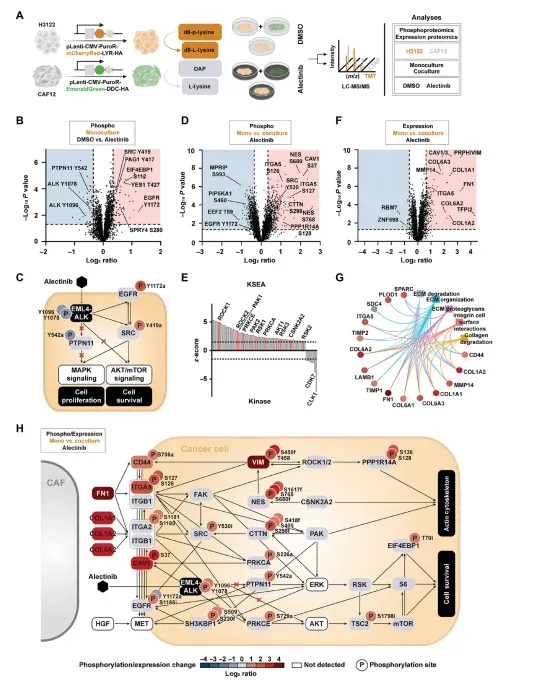
图3 EML4-ALK阳性非小细胞肺癌细胞与CAFs单培养及共培养中的磷酸化与表达蛋白质组学分析
功能验证环节,流式细胞术检测显示共培养使ITGB1激活水平提升2.4倍,而整合素拮抗剂cilengitide可将其恢复至单培养基线。在FN1包被的培养板上,H3122细胞对阿来替尼的敏感性下降50%,该效应被cilengitide完全消除。免疫印迹证实FN1通过激活FAK(Tyr397)磷酸化,进而恢复ERK、AKT及S6磷酸化,绕过ALK抑制(图4)。CRISPR-Cas9敲除ITGB1使共培养细胞的阿来替尼IC50值从1.2μM降至0.4μM,但未能完全消除耐药。当ITGB1敲除联合capmatinib处理时,共培养细胞的药物敏感性恢复至单培养水平。药理学实验复制了这一结果:cilengitide与capmatinib单用分别使共培养存活率降至68%和55%,而两药联用进一步降至25%,与单培养无统计学差异。值得注意的是,该联合策略在天然耐药的CUTO8与CUTO9细胞系中同样有效。
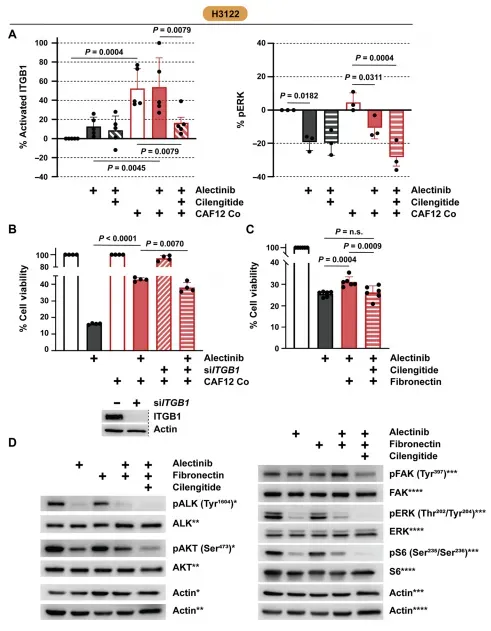
图4 靶向整合素信号对共培养介导的耐药性的影响
体内实验数据更具说服力。在mEA1同种移植瘤模型中,阿来替尼单药治疗4周后肿瘤体积为基线的60%,而阿来替尼+capmatinib+cilengitide三药联合组肿瘤体积降至基线的20%,相对单药组缩小67%(P<0.0001)。免疫组化显示,单药阿来替尼治疗后肿瘤组织S6(Ser235/236)磷酸化阳性面积比例反而升高至45%,提示代偿性激活;联合cilengitide后降至28%,三药联合进一步降至12%(图5)。临床数据挖掘揭示,在EGFR/ALK/ROS1突变阳性NSCLC队列中,FAP-α高表达与III-IV期肿瘤显著相关(P=0.036),而ITGB1高表达患者生存概率降低42%(HR=1.73,P=0.027),证实CAFs密度与整合素信号通路具有明确的临床预后价值。
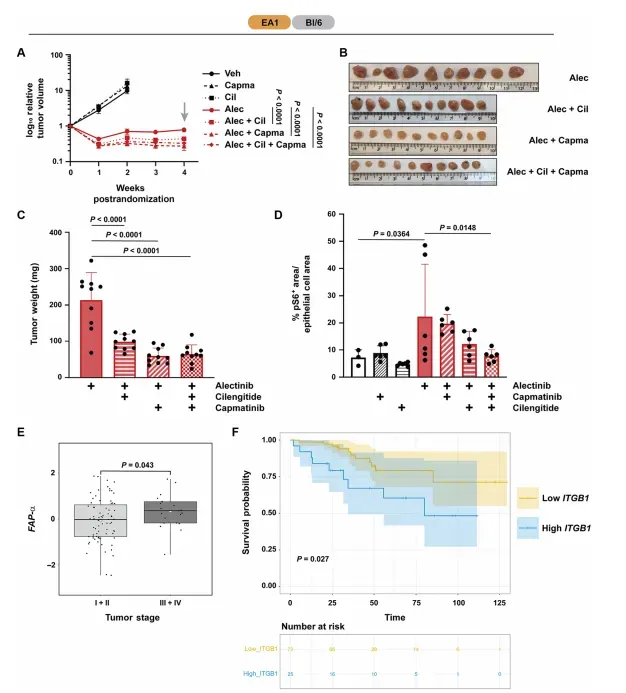
图6 ITGB1表达与靶向治疗在非小细胞肺癌肿瘤中的作用
总结本研究通过精巧的实验设计与创新的CTAP蛋白质组学技术,首次在单细胞分辨率层面系统阐明了CAFs介导EML4-ALK阳性NSCLC耐药的完整信号网络,突破性发现HGF-MET旁分泌轴与FN1-ITGB1近分泌轴的协同作用是耐药的核心机制。这一发现不仅解释了为何单独靶向MET或整合素仅能部分克服耐药的临床困惑,更提供了可立即转化的三药联合治疗方案。值得注意的是,该研究巧妙利用了部分ALK抑制剂(如lorlatinib、ceritinib)本身具有FAK/PYK2抑制活性的多靶点特性,提出可通过药物重定位策略实现"双通路阻断"而无需真正联用三种药物,为降低临床毒性风险提供了可行路径。
参考文献
HU Q, REMSING RIX L L, DESAI B, et al. Cancer-associated fibroblasts confer ALK inhibitor resistance in EML4-ALK–driven lung cancer by concurrent integrin and MET signaling[J]. Sci Signal, 2025, 18(1): eads7662.
编辑:白术
审核:薄荷
封面图源:CMT







