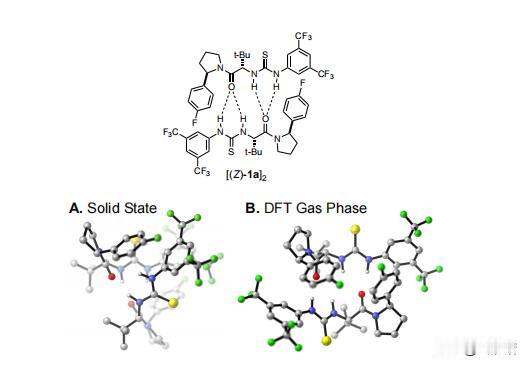
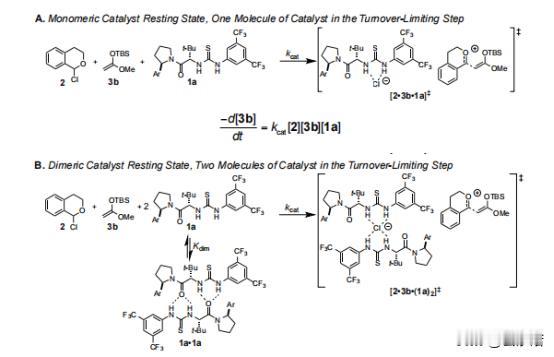
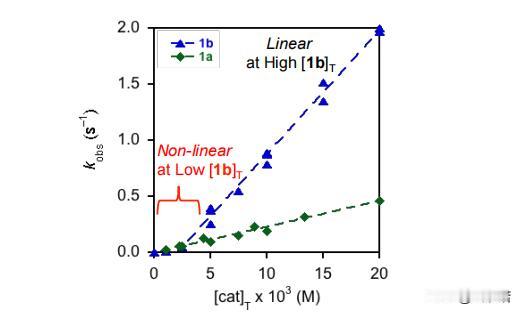
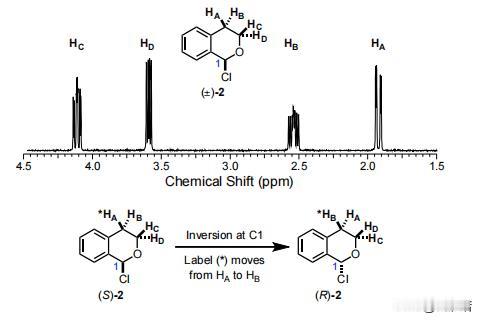
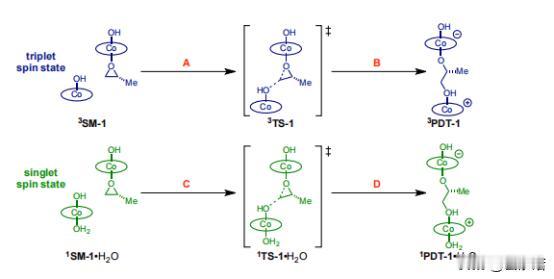
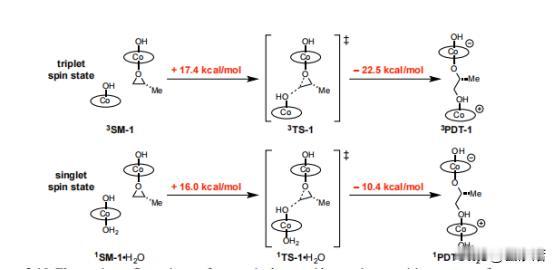
在HKR的反应中如何能够催化出(salen)Co-和(salen)Co-OH复合物 前言:在有机化学的领域中,分子间的相互作用和结构对反应性质产生了深远的影响,而在水解动力学分辨率(HKR)的反应中,(salen)Co-环氧复合物和反应性的(salen)Co–OH两种配位化合物扮演着十分关键的角色。 其中环氧化物络合物和亲核 (salen)Co-OH 络合物,在HKR(水解动力学拆分)过程中发挥着非常重要的作用,而(salen)Co-OH 配合物有两种建议的形式:六配位、低自旋构型(11b•H2O,S = 0)和五配位、中间自旋构型(31b,S = 1)。 要知道,六配位形式(11b•H2O)是专门负责HKR的亲核物质,这个物质在各种反应中都表现出了亲核的特性,其中也包括N-配位氨基酯配体的侧酯基团的水解,而六配位配合物11b•H2O 表现出具有明显阶梯构象的伪八面体几何形状。 五配位的配合物31b则采用扭曲的方锥体几何形状,值得注意的是,有相关计算表明,31b在五配位形式下保持稳定,并且对水的亲和力极小。 相比之下,11b•H2O 作为六配位的络合物则是最稳定的,这也表明它们与水的结合力很强,虽然说不同的理论方法的结果存在一些差异,但它们都出现了一致的趋势:能量最低的五配位配合物对应于三重态,而能量最低的六配位配合物对应于单重态。 如果对基态下各个Co(III)配合物进行检查之后,我们可以发现通过对 1,2-环氧丙烷(称为 TS-1)开环过渡结构的计算势垒进行比较,可以得出了一个有趣的现象,11b•H2O比31b表现出了更大的亲核性。 具体来说的话也就是,单重态流形中的环氧化物打开的势垒计算为比三重态流形计算的低 1.4 kcal/mol,而这些也恰好证明,六配位Co(III)配合物1 1b•H2O中的羟基配体确实表现出高度的亲核性。 在对计算出的过渡结构进行了分析之后,我们也为观察到的行为进行了解释:环氧化物开环的过渡态,会沿着反应坐标相对较早地发生,在这个阶段中,羟基配体仍然与Co(III)中心完全结合。 这也就意味着,亲核性与羟基配体的配位稳定性,甚至是不稳定性都没有直接的关系,反而是与钴原子结合时的亲核性质有关。 不管从哪个角度来看,我们这次的观察结果都挑战了之前的假设,因为这强调了催化活性不仅取决于配体未连接到金属中心时的行为,还取决于它们与钴配位时的行为。 需要值得注意的是,三线态流形的交叉在决定速率的环氧化物开环步骤之后,是会实现催化剂周转的。 为了能够更准确地解释B3LYP泛函往往会低估的色散相互作用,我们利用了Truhlar的M06-L 元GGA泛函对B3LYP优化的几何结构进行了单点计算,这些计算采用了更大的6-31+G(d,p) 基组,而这个基组以其在各种非共价相互作用基准中的强大性能而闻名。 不仅如此,在计算的过程中,我们还需要用到明尼苏达功能M05-2X,这个功能被认为可以准确预测非共价相互作用控制的反应中的催化剂结构-对映选择性关系,虽然这种方法有时会高估选择性趋势的幅度,但在这种情况下它仍然很有价值。 通过B3LYP和M06-L的计算,我们可以看到非常明显的趋势:改变(salen)Co(III)催化剂分子或环氧化物的绝对立体化学,会导致更高的过渡结构能,而这一发现强调,对于HKR反应十分重要的是必要立体化学匹配通过所选的 DFT 方法得到了很好的再现。 从本质上讲,这种计算分析支持了实验观察,重申了立体选择性在环氧化物开环反应中的重要性,而计算的能量和立体化学之间的相关性,还强调了分子排列和反应性之间复杂的相互作用,最终揭示了水解动力学拆分过程的立体选择性结果背后的驱动力。 总结:在这个实验中,我们通过计算分析,揭示了(salen)Co-环氧复合物和反应性的(salen)Co–OH复合物在反应中的重要作用,而且还发现计算方法在模拟非共价相互作用方面的应用,计算结果与实验结果具有一致性,为我们提供了有关分子间相互作用的更深入理解。 通过结合实验和计算,我们确认了这些关键化合物在环氧环开放反应中的作用,为有机化学领域的研究提供了新的洞察和启示,可以说,这一研究为设计和优化复杂有机反应,以及探索分子间相互作用在化学反应中的影响,提供了重要的指导和基础。